0
相关推荐



QIAseq Stranded RNA Library Kits provide a superior method for generating Illumina compatible RNA-seq libraries from total RNA or mRNA enriched samples. For applications such as gene expression, fusion gene or mutation detection, QIAseq Stranded mRNA Select Kits include an optimized mRNA enrichment protocol with all the reagents and components required to build high-quality RNA-seq libraries. QIAseq Stranded RNA Library Kits use a unique protocol, which does not require actinomycin D to retain strand specificity or dUTP to ensure stranded library construction, thereby ensuring highly sensitive detection of low-expression RNA molecules with increased complexity and transcript coverage. QIAseq Stranded RNA Library Kits use CleanStart HiFi PCR Mastermix to ensure high-fidelity amplification of libraries and to protect from PCR contamination. CleanStart HiFi PCR Mastermix is included in each kit or can be used separately with other library preparation methods. For one-step, rRNA and globin mRNA removal in just 20 minutes, use QIAseq FastSelect RNA Removal Kits.


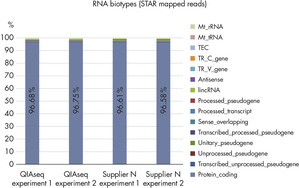
| QIAseq experiment 1 | QIAseq experiment 2 | |
| Reads mapped in pairs | 91.86% | 92.28% |
| Reads mapped in broken pairs | 4.84% | 4.47% |
| Reads not mapped | 3.29% | 3.25% |
| Total | 100 | 100 |
| % | QIAseq experiment 1 | QIAseq experiment 2 |
| Uniquely mapped reads | 92.02% | 91.69% |
| Reads mapped to multiple loci | 4.51% | 4.55% |
| Reads mapped to too many loci | 0.05% | 0.05% |
| Reads mapped: Too short | 3.38% | 3.68% |
| Reads mapped: Other | 0.04% | 0.04% |
| Total | 100 | 100 |
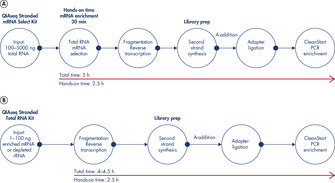
QIAseq Stranded RNA Library Kits enable fast, efficient and accurate NGS library construction from not only high-quality mRNA, but also low-quality fragmented total RNA of various origins. After an optional RNA fragmentation step, the reverse transcription step generates first strand cDNA.
Strand specificity is ensured due to optimized combinations of RT enzyme and buffer provided by QIAseq Stranded RNA Library Kits, removing the need to use toxic reagents such as actinomycin D. In the second strand synthesis step, a special combination of enzymes with carefully adapted buffer formulations allow the degradation of RNA, the generation of a second cDNA strand and the generation of blunt DNA ends and A-base addition necessary for efficient ligation of the Illumina compatible adapters. The special strand-specific ligation step of the QIAseq Stranded RNA Kit protocol ensures strand specificity without the need for additional reagents or laborious and time-consuming protocol steps. The included adapter plates with dual index technology can be used for the sequencing of multiple samples (up to 96). The CleanStart PCR mix efficiently amplifies the generated RNA-seq library irrespective of high GC or AT sequences, and also degenerates contaminating material such as previously generated NGS libraries. The paramagnetic QIAseq Beads included with kits provide fast and efficient reaction cleanup between protocol steps. High-quality RNA-seq libraries can be generated in just 4–5 hours.
The RNA fragmentation step using the reverse transcription buffer ensures the generation of RNA libraries with optimal insert sizes. The fragment size can be adapted by shorter or longer incubation times. The protocol also allows the use of input RNA that has already been fragmented, as well as heavily degraded low-quality FFPE RNA. In the reverse transcription step, optimized RT enzyme and buffer formulations ensure strand specificity during cDNA generation. Toxic reagents such as actinomycin D, which negatively affects reverse transcription efficiency, is not required.
The enzyme mix and buffer formulation are optimized to complete second strand synthesis, end-repair and A-addition in a very short time. First, RNA molecules are degraded. Next, cDNA molecules are used as templates to generate a second cDNA strand. Then, blunt double-stranded DNA is generated. Finally, an A-nucleotide is added to the 5' end of the cDNA. A modification of dexoynucleotides, which is needed with the dUTP method, is not required and ensures optimal efficiency of cDNA generation.
Strand-specific ligation of Illumina compatible adapters enables fast and efficient generation of RNA-seq libraries with cDNA prepared from the previous library preparation step. The strand-origin information of the initial RNA can be maintained without additional reagents, modified nucleotides or protocol steps. The included ready-to-use adapter plates allow the sequencing of up to 96 different samples by the combination of a unique 8-nucleotide i5 and i7 index barcode.
The QIAGEN proprietary CleanStart PCR Mix efficiently and uniformly amplifies the RNA-seq library regardless of the G/C content of the template, while also protecting against PCR contamination. The combination of an optimized buffer formulation and a Hotstart HiFi polymerase enzyme ensures low mutation rates and high processivity for the enrichment of an RNA-seq library. The CleanStart decontamination step allows the removal of contaminating material, such as previously generated NGS libraries, to maximize sample purity.
QIAseq Stranded RNA Library Kits generate RNA-seq libraries from low RNA amounts of various origins and species. The resulting highly complex and strand-specific NGS libraries, in combination with the decontaminating properties of the CleanStart PCR Mix, allow the analysis of even low-abundance transcripts with high sensitivity. The streamlined, 4–5 hour protocol allows the generation of NGS libraries, library QC measurements and the start of an NGS sequencing run in just one working day.
温馨提示:因厂家更改产品包装、产地或者更换随机附件等没有任何提前通知,且每位咨询者购买情况、提问时间等不同,为此以下回复仅对提问者3天内有效,其他网友仅供参考!若由此给您带来不便请多多谅解,谢谢!
服务热线
0771-3293894


 客服
客服