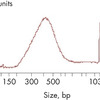
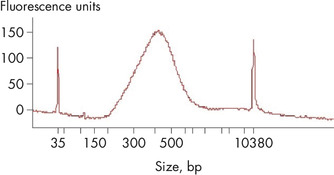
A portion of the 11 μl sequencing library was used as the starting material for the library QC and quantification. Library QC using an Agilent Bioanalyzer is shown with a peak at 425 bp. Real-time PCR-based methods provide an accurate quantification of complete RNA-seq libraries with full adapter sequences. As a result, QIAGEN’s QIAseq Library Quant Array Kit (cat. no. 333304) or Assay Kit (cat. no. 333314), which contains laboratory-verified forward and reverse primers together with a DNA standard, is highly recommended for accurate quantification of the prepared library. Use 1 nM RNA-seq libraries as input for the denaturation procedure to ultimately load 3 pM for the MiSeq (V3 kit) and 1.2 pM for the NextSeq.
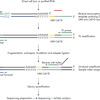
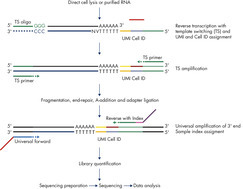
Library construction starts with lysis of cells or addition of total RNA to the 96- or 384-well plates that have the reverse transcription primers. Poly-A RNA is reverse transcribed using LNA-enhanced chemistry with an oligo-dt primer containing a random Unique Molecular Index (UMI) and a fixed cell ID. Following reverse transcription, cDNA is combined into a single tube for amplification, fragmentation, end-repair, A-addition and adapter ligation. During library amplification, up to 48 different sample indices can be assigned. The combination of cell IDs and sample IDs enables up to 18,432 libraries to be sequenced simultaneously.

The QIAseq UPX 3' Transcriptome Kit enables high-throughput next-generation sequencing (NGS) of polyadenylated RNAs. The kit is intended for library construction and analysis of single cells, cell pellets and ultra-low amounts of total RNA, starting with single cells, cell pellets or isolated RNA. QIAGEN’s Sample to Insight approach makes transcriptome and targeted gene expression accessible to researchers who demand high-quality results but do not have the time or experience in NGS workflow optimization or have the ability to create complicated bioinformatic pipelines for read alignment and differential gene expression/single-cell analysis.
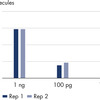
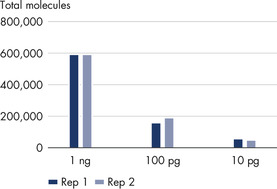
The QIAseq UPX 3’ Transcriptome Library Kit was used to construct libraries from 10 pg, 100 pg and 1 ng aliquots of HCT 116 total RNA. For each experimental replicate (n=2), reverse transcription and template switching were performed on eight aliquots of each RNA amount. Following this, each set of eight was combined into one tube, since each cDNA is tagged with a unique cell/well ID. Subsequent library construction steps were performed in a single tube and sequencing was performed on a MiSeq. QIAseq UPX 3’ analysis software was used for primary results mapping and deconvolution of the cell/well IDs. The number of captured molecules for each RNA amount are presented, demonstrating the reproducibility of the workflow.
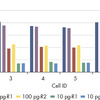
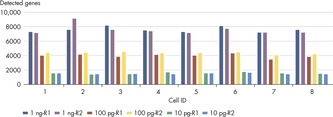
The QIAseq UPX 3’ Transcriptome Kit was used to construct libraries from 10 pg, 100 pg and 1 ng aliquots of HCT 116 total RNA. For each experimental replicate (n=2), reverse transcription and template switching were performed on eight aliquots of each RNA amount. Following this, each set of eight was combined into one tube, since each cDNA is tagged with a unique cell/well ID. Subsequent library construction steps were performed in a single tube and sequencing was performed on a MiSeq. QIAseq UPX 3’ analysis software was used for primary results mapping and deconvolution of the cell/well IDs. The number of detected genes from each aliquot of RNA (eight per RNA input amount) is presented, demonstrating the consistency of detection.





 客服
客服